
Atoms are the fundamental building blocks of matter, yet their behavior is governed by complex quantum mechanics that often defy simple prediction. Each atom consists of a positively charged nucleus surrounded by negatively charged electrons, and the interactions between these particles can lead to unpredictable behaviors, especially when they combine to form molecules. The challenge of accurately simulating these interactions has been a persistent hurdle for scientists, allowing only a limited understanding of molecular dynamics. Until recently, the computational power required to simulate these processes over long periods has far exceeded what was available, leaving many questions unanswered in fields ranging from material science to pharmacology.
The Traditional Approach: Schrödinger’s Equation
At the heart of quantum mechanics lies the Schrödinger equation, which is essential for determining the energy levels of quantum systems. However, solving this equation for molecules with more than a few atoms can turn into a formidable task, often requiring extensive computational resources and time. Traditional methods are not just slow; they’re also inefficient for anything but the simplest molecules. As a result, researchers have been forced to rely on approximations and less accurate models, which can lead to significant errors in the understanding of molecular properties and behaviors. The dream of achieving precise, long-term molecular dynamics simulations has seemed as elusive as ever.
The Emergence of Machine Learning
The glorious rise of machine learning (ML) in recent years has opened new avenues in computational chemistry. Machine learning offers a pathway to circumvent the complexities of quantum calculations by directly predicting molecular outcomes based on learned interactions. This paradigm shift allows researchers to harness vast amounts of data and uncover underlying patterns among electron interactions without the need for explicit calculations from Schrödinger’s equation. While the initial challenge lies in “teaching” algorithms to effectively model molecular interactions, the benefits could outweigh the hurdles.
A New Approach by BIFOLD and Google DeepMind
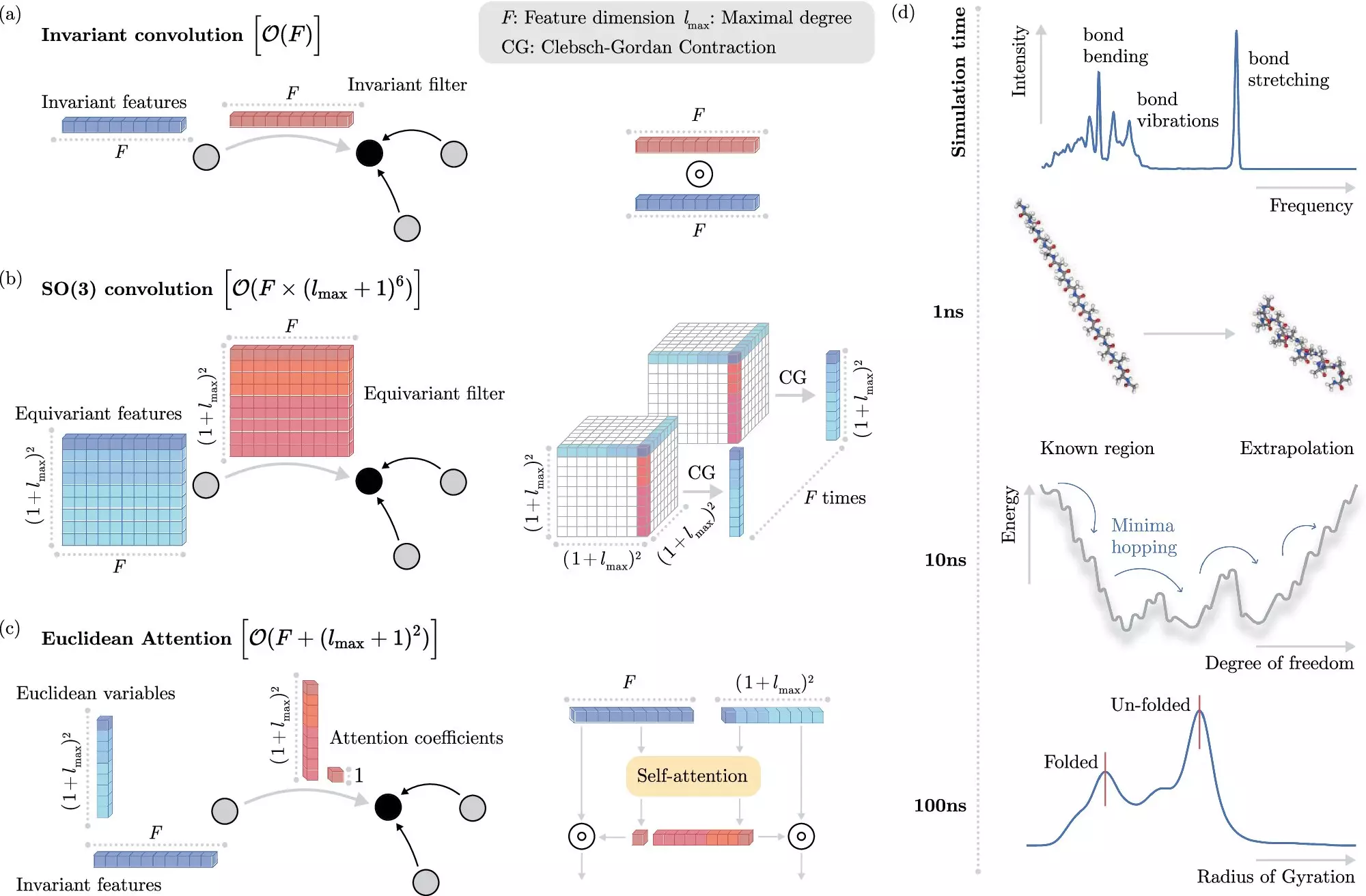
An inspiring collaboration between researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) and Google DeepMind has recently borne fruit in this realm. Their novel algorithm has introduced a transformative way of simulating molecular dynamics, enabling researchers to conduct simulations that were previously unimaginable. By decoupling invariances—properties of molecules that remain consistent despite spatial movement—the new algorithm optimizes computation to focus on the most relevant aspects of physical systems. Instead of relying on computationally intensive processes, this fresh approach streamlines the method to yield accurate results in significantly less time.
Efficiency Redefined: A Leap Forward
The implications of this algorithmic breakthrough are staggering. Tasks that once took months or even years on powerful supercomputers can now be completed in mere days on standard computing systems. This leap in efficiency reveals the potential of long-time scale simulations, integral for understanding complex chemical systems, from the binding interactions of proteins to the fundamental properties of new materials. As Dr. Stefan Chmiela from BIFOLD indicates, this significant advancement not only enhances scientific inquiry but also radically alters the landscape of molecular research altogether.
Applications Beyond Imagination
The implications extend beyond theoretical pursuits. For instance, the accurate simulation of molecular interactions opens the door to groundbreaking developments in drug discovery. By enabling accurate predictions of how new compounds interact with proteins in the human body, researchers can design drugs more effectively, circumventing traditional and often resource-intensive experimental methods. This advancement promotes not just scientific innovation but serves to enhance sustainability in research practices, reducing the need for extensive lab trials and potentially harmful materials.
Furthermore, the team’s applications of the algorithm demonstrated its efficacy in identifying more stable structural components from complex molecules, such as docosahexaenoic acid—critical for human brain health—by scanning through thousands of possibilities with unprecedented precision. This not only showcases the algorithm’s robustness but also its capacity for real-world application in health science and material engineering.
The Future of Molecular Simulations
As researchers look ahead, integrating advanced machine learning techniques with fundamental physical principles remains a critical frontier. The continued pursuit of algorithms capable of simulating larger, more complex systems, while accurately understanding long-range interactions, poses a tantalizing challenge. The combination of cutting-edge computational science and foundational chemistry promises a brighter, more efficient future for understanding the natural world, offering researchers new tools to decode the mysteries of matter. These innovations position the scientific community at the brink of monumental progress, which could redefine our relationship with everything from materials to health care.